Васильев А.О., Говоров А.В., Пушкарь Д.Ю.
Кафедра урологии ГБОУ ВПО Московский государственный медико-стоматологический университет имени А.И. Евдокимова Минздрава России, Россия, Москва
Адрес: 127206, Москва, ул. Вучетича д. 21, корп. 2, тел. 8(495)6113129 Эл.почта: alexgovorov@newmail.ru, alexvasilyev@me.com, pushkardm@mail.ru
Введение. Спектр врожденных аномалий почек и мочевых путей чрезвычайно широк и варьирует от легких и бессимптомных пороков, таких как удвоение мочеточника до тяжелых, подчас не совместимых с жизнью, таких как двусторонняя почечная агенезия или дисплазия. В результате тесной эмбриогенетической связи мочевой и половой систем человека аномалии развития органов мочевой системы (ОМС) в 33% случаев связаны с пороками развития половых органов, что в дальнейшем может приводить к развитию бесплодия. В общем числе поражений аномалии развития ОМС составляют в среднем 25% от общего числа всех генетических пороков, диагностируемых внутриутробно [1]. Врожденные пороки развития (ВПР) мочеполовой системы одна из самых многочисленных групп врожденных аномалий, включающих поражение почек, мочеточников, мочевого пузыря, уретры, а также мужских и женских гениталий.
О возможности одновременного развития врожденнойаномалии по чек и нижних мочевых путейбыло сообщено в руководстве Пытеля А .Я. и Голигорского С.Д. [2]. В 1998 году E. Yerkes и H. Nishimura был предложен термин CAKUT (congenital anomalies of the kidney and urinary tract) – врожденные аномалии почки и мочевого тракта. В своем исследовании авторы описали влияние ангиотензина в формировании врожденных аномалийпочек и мочевого тракта у мышей и у человека [3]. В настоящее время на долю CAKUT-синдрома приходится около 30% всех аномалий развития ОМС и до 50% всех случаев пальпируемой абдоминальной массы у новорожденных [4, 5]. Частота данного синдрома составляет 1 случай на 500 новорожденных [6].
CAKUT может быть как составнойчастью различных мультиорганных г енетических синдромов, так и самостоятельным поражением. В составе других генетических синдромов CAKUT описан приблизительно при 500 мультиорганных синдромах, как например, синдром почечного кистоза и сахарного диабета [7]. По данным Renkema K.Y. и соавт. до 10% изолированных форм CAKUT-синдрома могут быть сопряжены с наследственным фактором. В большинстве случаев семейных форм у ближайших родственников состояние протекает бессимптомно [8]. К синдрому CAKUT относят почечную дисплазию с/без гипоплазии, обструкцию мочевого тракта и пузырно-мочеточниковый рефлюкс (ПМР) (Таблица 1).
К основным причинам развития CAKUT-синдрома относят одновременную мутацию генов PAX2 и EMX2. Исследования показали отсутствие подобной мутации генов у здоровых эмбрионов мышей и у человека, причем у людей оба гена находятся на хромосоме 10 q и их мутация сопровождается полнойд еструкцией хромосомы [9]. Изучив роль PАХ2 в развитии ОМС, Chuary Ya. пришел к выводу, что отрицательное влияние на нефрогенез оказывает взаимоотношение исследуемого гена с такими транскрипционными факторами, как Gdnt, Ret, SHH, Wnt4, Fgt [10].
Таблица 1. Структура CAKUT-синдрома
| Почка |
Мочевые пути |
| Агенезия почки |
Агенезия – отсутствие треугольника мочевого пузыря |
| Дисплазия почки (в том числе кистозная и мультикистозная) |
Стеноз прилоханочного отдела мочеточника |
| Гипоплазия почки |
Мегауретер |
| Удвоение собирательной системы |
Задний клапан уретры |
| Подковообразная почка |
Пузырно-мочеточниковый рефлюкс |
Для полного понимания причин возникновения врожденных аномалий развития мы считаем целесообразным кратко изложить основные этапы эмбриогенеза ОМС [11].
Внутриутробное развитие человека продолжается в среднем 280 суток, в течение которых принято выделять три периода: начальный (1-я нед.), зародышевый (2-8-я нед.), плодный (с 9-й нед. развития до рождения). К концу зародышевого периода завершается закладка основных эмбриональных зачатков тканей и органов. В процессе эмбриогенеза человека последовательно закладываются три парных выделительных органа: передняя почка или предпочка (pronephros), первичная почка (mesonephros) и постоянная (окончательная) почка (metanephros).
Предпочка образуется из передних 8-10 сегментных ножек (нефротомов) мезодермы и состоит из эпителиальных трубочек, один конец которых слепо замкнут, а другой конец обращен в сторону сомитов, где канальцы, объединяясь, формируют мезонефральный (Вольфов) проток. У зародыша человека предпочка не функционирует в качестве мочеобразующего органа и вскоре после закладки подвергается обратному развитию. Мезонефральный проток сохраняется и растет в каудальном направлении.
Первичная почка формируется из большего числа нефротомов (до 25), расположенных в области туловища зародыша. С течением времени сегментные ножки отделяются от сомитов и спланхнотома и превращаются в слепые канальцы первичной почки. Канальцы растут по направлению к мезонефральному протоку и одним концом сливаются с ним. Навстречу к другому концу канальца первичной почки растут сосуды от аорты, которые распадаются на капиллярные клубочки. Каналец своим слепым концом обрастает капиллярный клубочек, образуя капсулу клубочка. Капиллярные клубочки и капсулы вместе формируют почечные тельца. Возникший при развитии предпочки мезонефральный проток открывается в заднюю кишку
Закладка окончательной почки происходит на 2-м мес. внутриутробного развития, но окончательное ее развитие завершается лишь после рождения ребенка. Эта почка образуется из двух источников мезонефрального протока и нефрогенной ткани. Последняя представляет собой не разделенные на сегментные ножки участки мезодермы в каудальной части зародыша. Мезонефральный проток растет по направлению к нефрогенному зачатку, и из него в дальнейшем формируются мочеточник, почечная лоханка с почечными чашками, а от последних возникают выросты, превращающиеся в собирательные протоки и трубочки. Эти трубочки играют роль индуктора при развитии канальцев в нефрогенном зачатке. Из последнего образуются скопления клеток, которые превращаются в замкнутые пузырьки. Разрастаясь в длину, пузырьки превращаются в слепые почечные канальцы, которые в процессе роста S-образно изгибаются. При взаимодействии стенки канальца, прилежащей к слепому выросту собирательной трубочки, происходит объединение их просветов. Противоположный слепой конец почечного канальца приобретает вид двуслойной чаши, в углубление которой врастает клубочек артериальных капилляров. Здесь формируется сосудистый клубочек почки, который вместе с капсулой образует почечное тельце. Образовавшись, окончательная почка начинает быстро расти и с 3-го мес. оказывается лежащей выше первичной почки, которая во второй половине беременности атрофируется.
Воздействие неблагоприятных факторов на организм беременной женщины в зародышевый период (с 4 по 8 нед.) может привести к агенезии почек и мочеточников, эктопии устья мочеточника, образованию дополнительного мочеточника со слепым концом или слабо развитой почкой.
Воздействие различных неблагоприятных факторов на 9-12 нед. развития может привести к формированию ретрокавального, ретроилиакального мочеточника, гетеролокальной дистопии почек и мочеточников, эктопии устья мочеточников, формированию сужений или клапанов различных отделов мочеточников, недостаточности складок слизистой устья мочеточников, врожденному недоразвитию или отсутствию мышечного слоя, высокому отхождению мочеточника от почечной лоханки.
В плодный период (с 13-й недели) возможно формирование извилистости мочеточника, его изгибов, укорочения, удлинения, дисбаланса сократительной функции, врожденной недостаточности иннервационного аппарата и дисплазии мочеточника [12].
Онтогенетические механизмы, лежащие в основе этиологии аномалий развития и CAKUT-синдрома являются многофакторными. Многие из них были получены путем вскрытия человеческих эмбрионов, другие – в эксперименте на животных. Подробная их характеристика представлена на рисунке 1 [5].
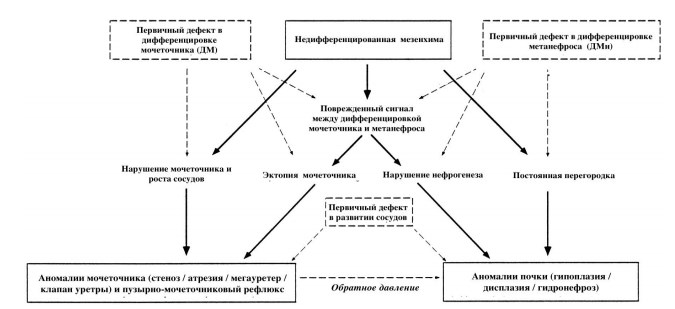
Рис.1. Онтогенетические основы формирования CAKUT-синдрома. Пунктирной стрелкой указана перекрестная аномалия.
Материалы и методы. Были проанализированы источники в англоязычной литературе, поиск которых проводили по базам данных Cochrane Library's, Medline (Pubmed, BioMedNet), Scopus и Biosis, используя ключевые слова «аномалии развития», «врожденные пороки развития», «CAKUT», «эмбриогенез органов мочевыой системы».
Обсуждение. Проявление врожденных пороков ОМС, в том числе CAKUT-синдрома, достаточно полно отражено в мировой литературе, однако данным аномалиям характерны особенности, которые до конца не изучены. Например, такие аномалии как стеноз лоханочно-мочеточникового сегмента (ЛМС), атрезия и мультикистозная дисплазия почки чаще всего являются односторонними; пренатальный гидронефроз новорожденных, связанный с ПМР и первичным мегауретером в подавляющем большинстве находят у мужчин [13, 14]. Некоторые из описанных аномалий часто развиваются одновременно [15]. Например, гипоплазия и дисплазия часто сопровождаются ПМР или стенозом ЛМС, атрезией ипсилатеральной или контралатеральной почки.
Самым распространенной врожденной аномалией ОМС является удвоение мочеточника, которое может быть полным и неполным. Неполное удвоение часто протекает вместе с раздвоением чашечно-лоханочной системы (ЧЛС). Большинство случаев протекает бессимптомно, однако риск инфицирования верхних мочевых путей возрастает в 20 раз [16]. Atwell J.D. и соавт. при обследовании 30 детей с неполным удвоением мочеточника пришли к выводу, что данная аномалия носит аутосомно-доминантный тип наследования, поскольку до 66% родственников первой степени родства имели такое же поражение [17].
Обструкция ЛМС является наиболее частой причиной антенатального гидронефроза и встречается с частотой 1 на 1000-1500 новорожденных [18]. Из всех известных причин развития данной аномалии большинством авторов высказывается предположение, что неполная мускуляризация мочеточника и чрезмерный апоптоз гладкомышечных клеток являются основными [19, 20].
В случае сращения нижних полюсов почек происходит образование подковообразной почки, расположенной в большинстве случаев ниже, чем обычно. В редких случаях наличие подковообразной почки связано с обструкцией ЛМС и инфицированием мочевых путей, гематурией и пальпируемой абдоминальной массой. Одним из наилучших вариантов лечения данной аномалии является трансперитонеальная лапароскопическая пиелопластика, позволяющая детально рассмотреть ЧЛС почки и наличие аномалий почечных сосудов [21].
Клапан задней уретры считается наиболее распространенной причиной обструкции нижних мочевых путей у новорожденных и эмбрионов мужского пола [22]. С данным пороком развития часто описывают синдром сливового живота, который по частоте встречаемости составляет 3,8 на 100 000 новорожденных мужского пола и характеризуется врождѐнным отсутствием, недостаточностью или гипоплазией мышц брюшной стенки в сочетании с мегацистисом, расширением мочеточников и простатического отдела уретры, а также двусторонним крипторхизмом. Одними из возможных методов лечения в данной ситуации является выполнение пузырно-амниотического шунтирования или выполнение везикоцентеза после рождения. Отличительной особенностью является характерный морщинистый вид передней брюшной стенки после операции, давший название этому синдрому [23, 24].
В большинстве случаев проявление обструктивной почечной дисплазии (в частности, мегацистис) может быть обнаружено при УЗИ плода, тогда как расширение мочеточника, чашечек или лоханки может быть заподозрено только при тщательном обследовании. Гистологическая картина может быть различной в зависимости от срока внутриутробной обструкции и включает сжатое мозговое вещество в результате избыточного гидростатического давления, создаваемого накопленной мочой в области малого таза [25]. К наиболее распространенным и изученным синдромам почечной дисплазии (группа цилиопатий) относят синдром Меккеля-Грубера, синдром Жубера, синдром короткого ребра, синдром Барде-Бидля, синдром асплении или полисплении и VACTER-L.
Синдром Меккеля-Грубера был впервые описан немецким биологом F. Meckel в 1822 году. Частота проявления у новорождѐнных составляет 1:9000. Синдром, как правило, приводит к летальному исходу в раннем детстве из-за тяжелой патологии почек и центральной нервной системы и характеризуется сочетанием поликистоза почек, аномалии развития центральной нервной системы (затылочное энцефалоцеле), фиброзных изменений печени и полидактилии [26] Синдром является генетически гетерогенным, аутосомно-рецессивным заболеванием. В настоящее время известно около 10 генов, мутации которых могут приводить к развитию синдрома, среди которых MKS1, TMEM216, TMEM67 и др.
Синдром Жубера редкая аутосомно-рецессивная патология, характеризующаяся полным отсутствием или недоразвитием червя мозжечка, умственной отсталостью, дистрофией сетчатки, глазной колобомой, нистагмом, полидактилией, кистозной почечной дисплазией и врожденным фиброзом печени. Синдром впервые описала M. Joubert в 1969 г. при обследовании четырех новорожденных в одной французско-канадской семье. Распространенность заболевания почек (кистозная почечная дисплазия и тубулоинтерстициальный нефрит) при синдроме Жубера и связанных с ними нарушений составляет в среднем 30% [27].
Синдром короткого ребра – генетическая патология, также относящаяся к группе цилиопатий и характеризующаяся наличием коротких рѐбер, укороченных конечностей, полидактилии и смертностью в раннем возрасте вследствие дыхательной недостаточности. Традиционно различают четыре подтипа этого синдрома: синдром Салдино — Нунана, синдром Маевского (Majewski syndrome), синдром Верма — Наумова (Verma-Naumoff syndrome) и синдром Бимер — Лангера (Beemer-Langer syndrome) [28]. Гистологическая картина почек схожа с таковой при синдромах Меккеля-Грубера и Жубера. Большинство генов, ассоциированных с синдромом короткого ребра, контролируют процесс интрафлагеллярного транспорта. Наиболее изучены мутации в гене NEK1 и TTC21B [29, 30].
Синдром Барде—Бидля является генетически гетерогенным заболеванием и встречается с частотой 1:120000 новорожденных. Характеризуется наличием как минимум четырѐх из шести первичных симптомов — ожирения, деградации сетчатки глаза, полидактилии, поликистоза почек, гипогонадизма и замедления умственного развития. К вторичными симптомам могут быть отнесены диабет, фиброз печени, атаксия, различные расстройства речи, асимметрия висцеральных органов, патология зубов, аносмия и потеря слуха. Известно 18 генов, мутации которых могут приводить к развитию синдрома [31].
В 1959 г. Ivemark B.L. и соавт. был описан вариант почечно-печеночноподжелудочной дисплазии в сочетании с аспленией [32]. В настоящее время в литературе отражено не более 10 сообщений, описывающих детей с данным аутосомно-рецессивным синдромом, характуризующимся мультикистозной дисплазией почки, печени и поджелудочной железы, недоразвитием легких, пороками сердца и в большинстве случаев приводящих к ранней младенческой смертности. На молекулярном уровне изучены два гена, мутация которых приводит к развитию данного состояния: NPNP3 и NEK8. Макроскопически отчетливо видна мультикистозная дисплазия почки [33].
Ассоциация VACTER-L — группа сочетанных аномалий развития, название которой составлено из первых букв пороков, входящих в состав синдрома [34]:
- V (Vertebral anomalies) — аномалии позвоночника, А (Anal аtresia) — атрезия ануса,
- С (Cardiovascular anomalies) — дефекты перегородок и другие пороки сердца,
- ТЕ (Tracheo-esophageal fistula) — трахеопищеводный свищ с атрезией пищевода,
- R (Renal defects) — аномалии почек, такие как агенезия, дисплазия, гидронефроз; единственная пупочная артерия.
- L (Limb defects) — дефекты лучевой кости — гипоплазия I пальца или лучевой кости, преаксиальная полидактилия и синдактилия. Большинством автором отмечается, что для постановки диагноза необходимо сочетание хотя бы трех из перечисленных симптомов. Описанное поражение встречается с частотой 1 на 10000—40000 новорожденных [14]
Выводы. Врожденные аномалии почек и мочевых путей группа состояний с различной степенью тяжести, многие из которых требуют междисциплинарного подхода для точной диагностики и улучшения последующего лечения. Учитывая, что многие из описанных врожденных аномалий являются наследственными, достижения в пренатальной диагностике, фетальной хирургии, а также целенаправленной терапии позволят улучшить прогноз и качество жизни новорожденных.
Литература
1. dos Santos Junior, A.C. Congenital anomalies of the kidney and urinary tract: an embryogenetic review / A.C. dos Santos Junior, D.M. de Miranda, Silva A.C. e Simões // Birth Defects Res C Embryo Today. – 2014, Vol. 102, №4 – P. 374-381.
2. Пытель, А.Я. Избранные главы урологии и нефрологии. Часть 1 / А.Я. Пытель, С.Д. Голигорский. Л.: Медицина, 1968. 312 с..
3. Yerkes, E. Role of angiotensin in the congenital anomalies of the kidney and urinary tract in the mouse and the human / E. Yerkes, H. Nishimura, Y. Miyazaki // Kidney Int. – 1998; Vol. 67, P. 75–77.
4. Toka, H.R. Congenital Anomalies of Kidney and Urinary Tract / H.R. Toka, O. Toka, A. Hariri, H.T. Nguyen // Samin Nephril. – 2010, Vol. 30, №4 – P. 374– 386.
5. Pope, J.C. IV. How they begin and how they end classic and new theories for the development and deterioration of congenital anomalies of the kidney and urinary tract, CAKUT / J.C. Pope IV, J.W. Brock III, M.C. Adams // JASN. – 1999, Vol. 10, №9 – P. 2018–2028.
6. Loane, M. EUROCAT statistical monitoring: identification and investigation of ten year trends of congenital anomalies in Europe / M. Loane, H. Dolk, A. Kelly // Birth defects Res A Clin Mol Teratol. – 2011, Vol. 91, №1 – P. 31-43.
7. Эрман, М.В. Нефрология детского возраста / М.В. Эрман. СанктПетербург: СпецЛит, 2010. 683 с.
8. Renkema, K.Y. EUCAKUT consortium/Novel perspectives for investigating congenital anomalies of the kidney and urinary tract (CAKUT) / K.Y. Renkema, P.J. Winyard, I.N. Skovorodkin // Nephrol. Dial. Transplant. – 2011, Vol. 26, №12 – P. 3843–3851.
9. Boualia, SK. Vesicoureter Reflux and Other Urinary Tract Malformation in Mice Compaund / S.K. Boualia, Y. Gartan, I. Murawski // PRoS One. – 2011, Vol. 6, №6 –P. 215–224.
10. Chuary, Ya. The role of PAX2 is regulation of kidney development and kidney diseases / Ya Chuary, // Ya Chion. – 2011, Vol. 33, №3 – P. 231– 238.
11. Афанасьев, Ю.И. Гистология, эмбриология, цитология. 6-е издание / Ю.И. Афанасьев, Н.А. Юрина. – М.: ГЭОТАР-Медиa, 2012. – 800 с.
12. Zwolińska, D. Genetics of congenital anomalies of the kidney and urinary tract / D. Zwolińska, D. Polak-Jonkisz, I. Makulska // Postepy Hig Med Dosw. – 2011, Vol. 15, №65 – P. 829-837.
13. Uehling, D.T. Urologic implications of the VATER association / D.T. Uehling, E. Gilbert, R. Chesney // J Urol. – 1983, Vol. 129, P. 352–354.
14. Solomon, B.D. VACTERL/VATER association / B.D. Solomon // Orphanet J Rare Dis. – 2011, Vol. 6, P. 6.
15. Atiyeh, B. Contralateral renal abnormalities in multicyctic-dysplastic kidney disease / B. Atiyeh, D. Husmann, M. Baum // J Pediatr. – 1992, Vol. 121, P. 6567.
16. Decter, R. Renal duplication and fusion anomalies / R. Decter // Pediatr Clin North Am. – 1997, Vol. 44, P. 1323–1341.
17. Atwell, J.D. Familial incidence of bifid and double ureters / J.D. Atwell, P.L. Cook, C. Howell // Arch Dis Child. – 1974, Vol. 49, P. 390–393.
18. Williams, B. Pathophysiology and treatment of ureteropelvic junction obstruction / B. Williams, B. Tareen, M.I. Resnick // Curr Urol Rep. – 2007, Vol. 8, P. 111–117.
19. Aoki, Y. Id2 haploinsufficiency in mice leads to congenital hydronephrosis resembling that in humans / Y. Aoki, S. Mori, K. Kitajima // Genes Cells. – 2004, Vol. 9, P. 1287–1296.
20. Kajbafzadeh, A.M. Smooth muscle cell apoptosis and defective neural development in congenital ureteropelvic junction obstruction / A.M. Kajbafzadeh, S. Payabvash, A.H. Salmasi // J Urol. – 2006, Vol. 176, P. 718–723.
21. Blanc, T. Laparoscopic pyeloplasty in children with horseshoe kidney / T. Blanc, E. Koulouris, N. Botto // J Urol. – 2014, Vol. 191, P. 1097–1103.
22. Quintero, R.A. In utero management of fetal lower urinary tract obstruction with a novel shunt: a landmark development in fetal therapy / R.A. Quintero, L.A. Gomez, L.A. Castro, C. Bermudez // J Matern Fetal Neonatal Med. – 2010, Vol. 23, P. 806–812.
23. Tonni, G. Prune-belly syndrome: case series and review of the literatura regarding early prenatal diagnosis, epidemiology, genetic factors, treatment, and prognosis / G. Tonni, V. Ida, V. Alessandro, M.P. Bonasoni // Fetal Pediatr Pathol. – 2013, Vol. 31, P. 13–24.
24. Granberg, C.F. Genetic basis of prune belly syndrome: Screening for HNF1β gene / C.F. Granberg, S.M. Harrison, D. Dajusta // J Urol. – 2012,Vol. 187, P. 272–278.
25. Yosypiv, I.V. Congenital anomalies of the kidney and urinary tract: a genetic disorder? / I.V. Yosypiv // Int J Nephrol. – 2012, Vol. 90, P. 83-90.
26. Opitz, J.M. Annals of morphology. Meckel on developmental pathology / J.M. Opitz, R. Schultka, L. Göbbel // Am J Med Genet. – 2006, Vol. 140A, P. 115– 128.
27. Joubert, M. Familial agenesis of the cerebellar vermis. A syndrome of episodic apnea, abnormal eye movements, ataxia, and retardation / M. Joubert, E. JeanJacques, J.P. Robb, F. Anderman // Neurology. – 1969, Vol. 19, P. 813–825.
28. Dagoneau, N. DYNC2H1 mutations cause asphyxiating thoracic dystrophy and short rib-polydactyly syndrome, type III / N. Dagoneau, M. Goulet, D. Geneviève, Y. Sznajer, J. Martinovic, S. Smithson, C. Huber, G. Baujat, E. Flori, L. Tecco, D. Cavalcanti, A.L. Delezoide, V. Serre, M. Le Merrer, A. Munnich, V. CormierDaire // Am J Hum Genet. – 2009, Vol . 84, №5 – P. 706-711
29. Thiel, C. NEK1 mutations cause short-rib polydactyly syndrome type Majewski / Thiel, A. Giessl //Am J Hum Genet. – 2011, Vol. 88, P. 106–114.
30. Davis, E.E. TTC21B contributes both causal and modifying alleles across the ciliopathy spectrum / E.E. Davis, Q. Zhang, Q. Liu // Nat Genet. – 2011, Vol. 43, P. 189–196.
31. Green, J.S. The cardinal manifestations of Bardet–Biedl syndrome, a form of Laurence–Moon–Biedl syndrome / J.S. Green, P.S. Parfrey, J.D. Harnett // N Eng J Med. – 1989, Vol. 321, P. 1002–1009.
32. Ivemark, B.L. Familial dysplasia of kidneys, liver and pancreas: a probably genetically determined syndrome / B.L. Ivemark, V. Oldfelt, R. Zetterstrom // Acta Paediatr. – 1959, Vol. 48, P. 1–11.
33. Frank, V. Mutations in NEK8 link multiple organ dysplasia with altered Hippo signaling and increased c-MYC expression / V. Frank, S. Habbig, M.P. Bartram // Hum Molec Genet. – 2013, Vol. 22, P. 2177–2185.
34. Uehling, D.T. Urologic implications of the VATER association / D.T. Uehling, E. Gilbert, R. Chesney // J Urol. – 1983, Vol. 129, P. 352–354.












Комментарии